Development News Brief
Get Galaxy! http://bitbucket.org/galaxy/galaxy-central/wiki/GetGalaxy
- new:
% hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist - upgrade:
% hg pull -u -r 50e249442c5a
Key Upcoming Galaxy Events
May 24-26, 2011 Galaxy Community Conference, Lunteren, The Netherlands.
- http://galaxy.psu.edu/gcc2011/
- April 24 is the Early registration deadline (save 20%!)
- http://galaxy.psu.edu/gcc2011/Register.html
April 13-14, NBIC Galaxy Hackathon, Belgium. Please submit your suggestions!
What's New
Workflow API
An API for executing workflows has been added.
Usage examples
/scripts/api/workflow_execute.py
/scripts/api/example_watch_folder.py.
Move Data Library Items
We have introduced a feature that enables you to move library datasets or folders (including all folder contents) to other locations within the same data library or to a different data library altogether.
Galaxy administrators can perform this feature on any data library item and can move items to any data library. Users that are not Galaxy administrators must be given the modify library item permission on an item in order to move it, and they'll need the add library item permission on the desired target data library folder in order for it to be displayed in the select list of targets.
Moving a single dataset
To move a single dataset, select the Move this dataset option from the dataset's pop-up menu.

The default behavior is to move items to other locations within the same data library, so you'll initially be presented with a list of valid folders from which to choose for the new target location.
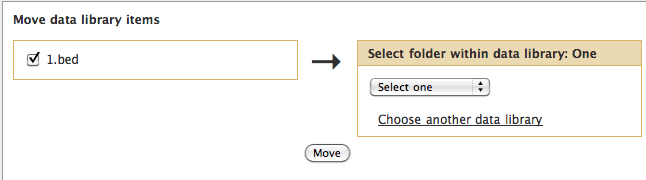
After selecting the desired folder, clicking the Move button will move the dataset to the selected folder.
Moving a Folder
Moving a folder is very similar to moving a single dataset - just select the Move this folder option from the folder's pop-up menu.

To move the folder to a different data library, after selecting the Move this folder option, click the Choose another data library link on the Move data library items page. The folders select list will change to a select list of data libraries to which you are authorized to move the item. When you select a target data library, the select list will change again to display the list of folders in the selected data library to which you are authorized to move the item. Clicking the Move button will move the folder and all of it's contents to the selected target folder within the selected target data library.
The target folders select list is filtered to include only valid folders to which you can move the item. For example, you cannot move a folder to one of it's own sub-folders in one step. To do this, the sub-folder must be moved outside of it's parent, and then the parent can be moved to the folder that it previously contained.

Updated & Improved
Data Content
-
Genomes
-
New & included in NGS Tools
- Saccharomcyes cerevisiae: Saccharomcyes_cerevisiae_S288C_SGD2010
- Arabidopsis lyrata: Araly1
- Purple Sea Urchin: strPur3 and Spur_v2.6
- Hydra: Hydra_JCVI
- Zebrafish: danRer7
- Poplar: Ptrichocarpa_156
- Chimpanzee: panTro3
- Northern White-Cheeked Gibbon: nomLeu1
- Korean Man AK1: Homo_sapiens_AK1
-
New & not included in NGS Tools
- Caenorhabditis remanei: caeRem2
-
Existing genomes added to NGS Tools
- hg19 Canonical female (no Y chromosome)
- Streptococcus pneumoniae R6: 278
- Drosophila virilis: droVir3 and droVir2
-
-
New LiftOver Files
- caeRem2 --> caePb1, caeRem2 --> caeRem3, caeRem2 --> cb3, caeRem2 --> ce4, caeRem2 --> priPac1, calJac3 --> hg18, canFam2 --> monDom5, danRer6 --> danRer7, danRer7 --> fr2, danRer7 --> gasAcu1, danRer7 --> hg19, danRer7 --> mm9, danRer7 --> oryLat2, danRer7 --> panTro3, danRer7 --> tetNig2, danRer7 --> xenTro2, droVir3 --> droVir2, fr2 --> danRer7, gasAcu1 --> danRer7, hg18 --> calJac3, hg19 --> danRer7, mm9 --> danRer7, panTro3 --> danRer7, panTro3 --> hg19, ponAbe2 --> calJac3, ponAbe2 --> monDom5, strPur2 --> ci2, tetNig2 --> danRer7, xenTro2 --> danRer7
-
Add Genomes to Your Instance
-
Current Galaxy Main Genomes
Current Tools
- Add more verbose error reporting to FASTQ Groomer tool. Provides more information to allow users to determine what is wrong with FASTQ files with invalid format.
- Enhance Bowtie wrapper to accept non-Sanger variant FASTQ files.
- Allow Upload Tool to function on
httpsURLs. -
Add count GFF features tool and tests:
- Filter and Sort --> GFF --> Filter GFF file by feature count using simple expressions.
- Tool counts the number of features in a GFF file. Note: this is different than the number of lines because a single GFF feature can often span multiple lines.
-
Tophat v1.2.0 support:
- (a) allow indel search.
- (b) max insertion and max deletion lengths.
- (c) library type.
- Updated
gff_filter_by_feature_counttool now accepts and correctly handles all GTF, GFF, and GFF3 files. - Changes for detecting and loading BAM data type Samtools version 0.1.13 or newer produces an error condition when attempting to index an unsorted BAM file. To determine if a BAM file is sorted, we first use Samtools to check the headers. If this does not provide a definitive answer and Samtools version 0.1.13 or newer is being used, we index the BAM file to see if it produces the error. This process provides a more robust approach to determining if the BAM file is sorted.
New Tools
- Multiple Alignments: ClustalW multiple sequence alignment program for DNA or proteins.
-
Motif Tools: Sequence Logo generator for FASTA data (example: ClustalW alignment).
- Both tools originated from the Community Tool Shed (see below).
- The Sequence Logo tool uses Weblogo3 wrapped into Galaxy to generate a sequence logo. The input file must be a FASTA file in your current history. It is recommended for viewing multiple-sequence alignment output from the ClustalW tool. Set the ClustalW output to FASTA to create the input for this tool.
A typical output looks like this:
Community Tools (Tool Shed)
- Tuning: Clarified what is being searched in the Tool Shed.
- New ClustalW wrapper (see http://www.clustal.org) for protein/dna multiple alignments based on the Galaxy ClustalW wrapper posted by Hans-Rudolf Hotz in an email on the developer list
- New Weblogo3 wrapper (http://weblogo.berkeley.edu) that creates sequence logos from FASTA data such as the output from a ClustalW alignment.
Data Libraries
- Disabled problematic eager loading on data libraries. Very large data libraries will load two to three times quicker.
- Upload Improvement with example walk-through:
Uploading data library datasets
- Apply a corrected version of the patch from Peter Cock that flips the "Preserve directory structure?" setting when uploading library datasets from filesystem paths. The checkbox is now "yes" instead of the original "no", and is checked by default.

- Flip the behavior of the "Copy data into Galaxy" feature when uploading library datasets (similar to 1 above) to be more clear and logical. Instead of a single checkbox, this is now a select list with clearly defined options. The default setting is to copy the files into the configured Galaxy file store.

- Allow importing items from a history to replace a library dataset with a new version. Previously, you could only replace a library dataset with a new version by uploading a single file.

Workflows
- Users can now import copies of their own Workflows and Histories.
Trackster
-
Enhancements:
- Implemented a data manager for Trackster and drawing is now completely tracked and controlled.
- Put show_insertions and show_differences in read track
config. -
Filtering now supports:
- (a) score columns in BED and GFF.
- (b) GTF attributes.
- Added BED and GFF support to visual analytics framework, and enable GOPS intersect and subtract tools to work with visual analytics.
-
Improvements:
- Insertions and deletions now shown in reads.
- Save and restore mode for feature and read tracks.
- Bug fixes for drawing feature tracks in
SquishandDensemode. - Menus and menu items now work correctly.
- Remove form from navigation controls so that enter key works properly and sets the chrom/low/high.
- Enable full keyboard navigation via arrow keys.
-
Bug fixes:
-
Fix issues with navigation input:
- (a) arrow keys no longer perform navigation
- (b) invalid chromosome names are handled well.
- Fix feature track bugs so that intervals are correctly drawn as half-open.
- Use gap to correctly position read connector in
Packmode. - Fix read id bug in Trackster BAM data provider.
-
Example of Trackster visualization:
Track of reads mapped using Tophat (spliced) including the transcript assembled from those reads.

User Interface (UI)
- SGD Yeast genome added as a Gbrowse display site.
- IGV added as an external display application. This is not enabled on Galaxy main, but is available for local instances.
CloudMan
FTPdata upload enabled. http://bitbucket.org/galaxy/galaxy-central/wiki/cloud
Source
- Improvements in reliability and speed regarding manipulation of instance data volumes.
- The value set in '
new_file_path' inuniverse_wsgi.iniwill now be used for creation of all temporary files. This obviates the need for setting$TEMPwhen running data source tools on the cluster. -
Datatypes:
- Add a '
subclass' flag to datatype definitions indatatypes_conf.xmlthat allows dynamic creation of a subclass. - Allow composite datatype datasets to be populated in the Upload tool from files that were uploaded to the Galaxy FTP server.
- Add a '
-
Tool configuration enhancements:
- Allow
DataToolParameterto be filtered on attributes other thandbkey(dynamic options). - Allow
FromParamToolOutputActionOptionandParamValueToolOutputActionOptionFilterto access attributes of parameter values.
- Allow
Tool Test Framework
- Specifying '
ftype="bam"' as a parameter on a test's<output>tag will now cause the test framework to use 'samtools view' to convert the file to SAM. This should make it easier to debug why the test tool output differs from your baseline test output.
Test framework enhancements:
- Allow
toolbox teststo upload a file found located insubdirectories. - Fix a bug occurring on the determination of uploaded dataset name during the handling of the removal of
.gzipor.zipextension from the uploaded filenames. - When using
re_match comparison methodin functional tests and line counts do not match, print out first 40 lines of the history file.
Admin Menu
- Add a checkbox to the Create Group page that if checked will create a new
Rolewith the same name. This provides a similar feature the the existing checkbox on the Create Role page. - However, the behavior is now changed such that new associations are created when the checkbox is checked, whereas before, only the
GrouporRoleobjects with the same name were created, but not associated with anything.
Bug Fixes
- Following the login link from the Logout page no longer redirects you to the Logout page once you have logged in.
- The peek setting method,
set_peek(), should now function consistently across datatypes descending fromText, particularly with respect to line count estimation. - Apply patch from Ry4an Brase to correct "NoneType preference on the jobs view" issue.
- Don't alter the contents of a file while uploading to a data library if using one of the Upload files from file system paths or Upload a directory of files options in conjunction with the Link to files without copying into Galaxy option. This partially resolves the issue where a supposedly sorted BAM file was being resorted upon upload to a data library when using this option. A better implementation of determining whether a BAM file has been sorted (so that it does not get resorted) remains to be done.
-
Extract Genomic DNA tool:
- Do not fix strand for GFF input.
- Handle non-GFF files when interpret features is
true.
- Set default value for Cuffdiff's minimum alignment count parameter to reflect v0.9.3.
- Enhance GFF reader to handle headers and comments and fix bug so that new feature starts when
transcript_idorgene_idchanges. - Suppress most logging for Cufflinks and Cuffdiff as the logging can generate output that is incompatible with
UTF-8 formatused by database. - Set message type to error when history deletion fails due to sharing.
- Show masthead links to Published Visualizations and Published Pages only if Tracks or Pages are enabled.
- Shared visualizations work again.
- Fix bug in
recently used tool submenu. - Change default value for Tophat coverage search to
true.
About Galaxy
The Galaxy team is a part of BX at Penn State, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State, and Emory University.
Join us at Twitter
#usegalaxy
http://twitter.com/#!/search/galaxyproject