On this page


This is the workshop page for the Tool Development from bright idea to toolshed - Data Managers Training Day session at GCC2014.
This workshop is offered on June 30, from 3:30 through 6pm, in Room 304 of Charles Commons.
Presenters: Dan Blankenberg and Jim Johnson
Summary
Galaxy tools can require installed reference data in order to be used effectively. For example, Bowtie requires prebuilt indexes in order to efficiently map sequences to a genome. Data Managers enable a Galaxy administrator to add reference data to a Galaxy server via the admin webpage or through the Galaxy API. This session covers the tool and ToolShed requirements for using reference data within galaxy tools, and the design and development of tool data managers to install reference data on a Galaxy server.
Helpful links
- https://galaxyproject.org/Admin/Tools/data-managers
- https://galaxyproject.org/admin/tools/tool-config-syntax
- For a video overview on Data Managers, see this presentation from GCC2013.
Prerequisites
- The virtual machine image for this workshop should be installed before you arrive.
- A wi-fi enabled laptop with a modern web browser. Google Chrome, Firefox and Safari will work best.
- Knowledge and comfort with the Unix/Linux command line interface and a text editor. If you don’t know what cd, mv, rm, mkdir, chmod, grep and so on can do then you will struggle in this workshop.
- Secure Shell (SSH) client software such as PuTTY for Windows, or the Terminal Application that comes with Mac OS.
15:30-15:40 Introduction
- Introduce presenters and circulating tutors
- Scope of class
15:40-16:00 Using Reference Data in Galaxy Tools
-
Graphical Overview of Interplay between Built-in Data and Galaxy Tools
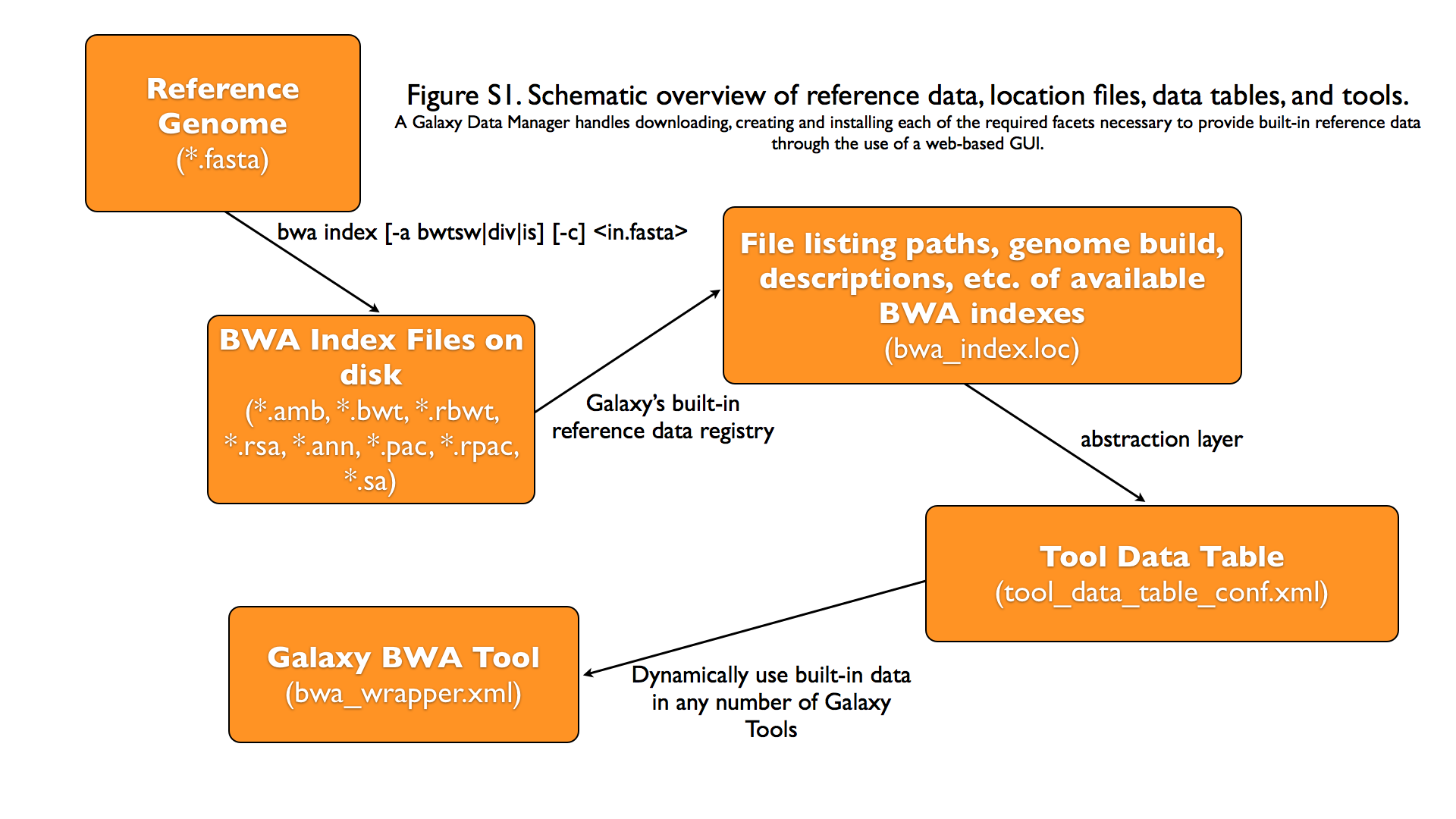
-
Discussion of *.loc files
- Used as a way to provide additional configuration details to a tool, without having to manually edit the actual tool XML file.
- Colloquially referred to as “loc” or “location” files.
- Often used to store the path (location on disk) of reference data and indexes, along with appropriate metadata (display names, dbkeys/genome builds)
- Need not end in the suffix of “.loc”, although they commonly do by convention.
- Tab delimited flat files, where each row is an entry in the table.
- Should not be accessed directly in a tool. The Tool Data Tables abstraction layer should be used.
-
Example of typical use of reference data in a Galaxy tool
- search toolshed for bwa tool that uses reference data
- Click “Repository Actions” and select “Browse repository tip files”
- Find and click on “bwa_wrapper.xml” and note the section that uses indexed data:
- search toolshed for bwa tool that uses reference data
<conditional name="genomeSource">
<param name="refGenomeSource" type="select" label="Will you select a reference genome from your history or use a built-in index?">
<option value="indexed">Use a built-in index</option>
<option value="history">Use one from the history</option>
</param>
<when value="indexed">
<param name="indices" type="select" label="Select a reference genome">
<options from_data_table="bwa_indexes">
<filter type="sort_by" column="2" />
<validator type="no_options" message="No indexes are available" />
</options>
</param>
</when>
<when value="history">
<param name="ownFile" type="data" format="fasta" metadata_name="dbkey" label="Select a reference from history" />
</when>
</conditional> 3. Find and click on "tool_data_table_conf.xml.sample" which defines the mapping to the "tool-data/bwa_index.loc" file <tables>
<!-- Locations of indexes in the BWA mapper format -->
<table name="bwa_indexes" comment_char="#">
<columns>value, dbkey, name, path</columns>
<file path="tool-data/bwa_index.loc" />
</table>
</tables> 4. Find and click on "bwa_index.loc.sample" #This is a sample file distributed with Galaxy that enables tools
#to use a directory of BWA indexed sequences data files. You will need
#to create these data files and then create a bwa_index.loc file
#similar to this one (store it in this directory) that points to
#the directories in which those files are stored. The bwa_index.loc
#file has this format (longer white space characters are TAB characters):
#
#<unique_build_id> <dbkey> <display_name> <file_path>
#
#So, for example, if you had phiX indexed stored in
#/depot/data2/galaxy/phiX/base/,
#then the bwa_index.loc entry would look like this:
#
#phiX174 phiX phiX Pretty /depot/data2/galaxy/phiX/base/phiX.fa-
start galaxy
open a terminal/shell and go to galaxy dir
cd /home/galaxy/Desktop/Data_Managers/galaxy/galaxy-central start a virtual env
. galaxy_env/bin/activateStop Galaxy if it’s running
sh run.sh --stop-daemonRestart Galaxy
sh run.sh --daemonWatch the Galaxy log
tail -f paster.log- Check that your galaxy instance is running. In your VM web browser, visit http://localhost:8080. Register your admin email address if you haven’t already done so and log in.
- You can also use the preconfigured username:
admin@galaxyproject.orgpassword:galaxy
- You can also use the preconfigured username:
- If you don’t have an admin menu item on your tool bar, refresh your browser. If the issue persists: adjust universe_wsgi.ini by adding an admin_user email you will register with when you first log in - use commas ONLY - no spaces - to separate admin email addresses. Then restart galaxy.
$ grep admin_users universe_wsgi.ini
admin_users = jj@msi.umn.edu- Install the BWA Galaxy tool
- From the Galaxy admin page: Click “Search and browse tool sheds”
- Click “Galaxy main tool shed”
- Searh for “bwa”
- Select “bwa_wrappers” owned by devteam
- Click “Install to Galaxy”
- Click “Install”
- In your terminal window:
- View shed_tool_conf.xml it should now contain a bwa_wrapper entry
- View shed_tool_data_table_conf.xml should have the bwa tool_data_table_conf.xml.sample table entries added.
- There should be a tool-data/bwa_index.loc file (copied from “bwa_index.loc.sample” if not already created)
- Upload some sample FASTQ datasets:
- http://www.bx.psu.edu/~dan/examples/gcc2014/data_manager_workshop/fastq/SRR507778-10k_1.fastqsanger
- http://www.bx.psu.edu/~dan/examples/gcc2014/data_manager_workshop/fastq/SRR507778-10k_2.fastqsanger
- These are paired end datasets created using Illumina technology, obtained from EBI SRA, and decreased to ~10,000 reads.
- When uploading these datasets set the datatype to “fastqsanger”.
- When the BWA tool is installed, Click “Analyze Data” menu item and select the BWA tool
- Note the choices available under “Select a reference genome:” hint: this list is empty
- Add a new built-in reference dataset, we will be using the sacCer1 genome build.
- Get the reference genome in the FASTA format. From your Galaxy root (galaxy@gcc2014:~/Desktop/Data_Managers/galaxy/galaxy-central$):
cd tool-data/mkdir sacCer1mkdir sacCer1/seqcd sacCer1/seqwget http://www.bx.psu.edu/~dan/examples/gcc2014/data_manager_workshop/sacCer1/sacCer1.fa
- Create BWA indexes for the reference genome.
- Download and compile BWA (http://bio-bwa.sourceforge.net/)
- or you can download and use a precompiled version:
cd ~/Desktop/Data_Managers/galaxy/galaxy-central/tool-data/sacCer1mkdir bwa_indexmkdir bwa_index/sacCer1cd bwa_index/sacCer1wget http://www.bx.psu.edu/~dan/examples/gcc2014/data_manager_workshop/bwa/bwachmod +x bwaln -s ../../seq/sacCer1.fa sacCer1.fa./bwa index sacCer1.fals -lah
- edit the tool-data/bwa_index.loc file adding a new entry
cd ~/Desktop/Data_Managers/galaxy/galaxy-central/tool-data/nano bwa_index.loc- add this entry to the end of the file:
sacCer1 sacCer1 S. cerevisiae Oct. 2003 (SGD/sacCer1) (sacCer1) /home/galaxy/Desktop/Data_Managers/galaxy/galaxy-central/tool-data/sacCer1/bwa_index/sacCer1/sacCer1.fa- Check the BWA tool for the new entry in your web browser
- Restart your galaxy server
- Again check the BWA tool for the new entry in your web browser
- If the your new entry does not appear, did you remember to separate the fields with TAB characters
- Align your FASTQ reads using the BWA tool to the newly added built-in reference genome data.
16:00-16:10 Introduction to DataManagers
-
The problem
- Administrator needed to know how to update each type of reference data
-
Data Managers to the rescue
- Allows for the creation of built-in (reference) data
- underlying data
- *.loc files
- data tables
- Specialized Galaxy tools that can only be accessed by an admin
- Defined locally or installed from the Tool Shed
- Flexible Framework
- not just Genomic data
- Interactively Run Data Managers through UI
- Workflow compatible
- Can Run via Galaxy API
- Allows for the creation of built-in (reference) data
-
How this operates within galaxy
- https://wiki.galaxyproject.org/Admin/Tools/DataManagers/HowTo/Define
- configuration
- universe_wsgi.ini
- data_manager_conf.xml
- shed_data_manager_conf.xml
16:10-16:30 Install a DataManager from the ToolShed
- https://wiki.galaxyproject.org/Admin/Tools/DataManagers/HowTo/Define
- configure your galaxy server to use Data Managers
- In your “universe_wsgi.ini” file these settings exist in the
[app:main]section:
- In your “universe_wsgi.ini” file these settings exist in the
# Data manager configuration options
enable_data_manager_user_view = True
data_manager_config_file = data_manager_conf.xml
shed_data_manager_config_file = shed_data_manager_conf.xml
galaxy_data_manager_data_path = tool-data-
Where enable_data_manager_user_view allows non-admin users to view the available data that has been managed.
-
Where data_manager_config_file defines the local xml file to use for loading the configurations of locally defined data managers.
-
Where shed_data_manager_config_file defines the local xml file to use for saving and loading the configurations of locally defined data managers.
-
Where galaxy_data_manager_data_path defines the location to use for storing the files created by Data Managers. When not configured it defaults to the value of tool_data_path.
-
From the Galaxy admin page:
- install data_manager_fetch_genome_all_fasta from Galaxy main tool shed
-
view in galaxy file system where the various elements land
- shed_data_manager_conf.xml
- shed_tool_data_table_conf.xml
-
from galaxy admin panel use data_manager_fetch_genome_all_fasta to install data
- Click on “Manage local data” Run Data Manager Tools Reference Genome - fetching View Data Manager Jobs Reference Genome - fetching View Tool Data Table Entries all_fasta
- Click on “all_fasta” under “View Tool Data Table Entries”
- You should see the current contents of tool-data/all_fasta.loc
- Click on “Reference Genome” under “Run Data Manager Tools”
- The Reference Genome tool form from data_manager_fetch_genome_all_fasta is displayed
- From the DBKEY to assign to data: list choose: sacCer2
- Click Execute
- Under the “User” menu select “Saved Histories”
- Select history: “Data Manager History (automatically created)”
- You should see your “Reference Genome” job in the history
- Go back to the Data Manager view by click on “Manage local data”
- Click on “all_fasta” under “View Tool Data Table Entries”
- You should see sacCer2 added to all_fasta
16:30-17:10 Create a DataManager
Create a local data manager to generate BWA indexes
We will need to:
- Create the Data Manager Galaxy tool
- Configure Galaxy to use this Data Manager tool
- Create the Data Manager Galaxy tool hint1: The Bowtie2 data manager is almost identical https://testtoolshed.g2.bx.psu.edu/view/jjohnson/data_manager_bowtie2_index_builder hint2: Dan has already done a bwa data manger http://toolshed.g2.bx.psu.edu/view/devteam/data_manager_bwa_index_builder
- highlight extra requirements/tags/attributes vs regular galaxy tool
Data Manager Tool
A Data Manager Tool is a special class of Galaxy Tool. Data Manager Tools do not appear in the standard Tool Panel and can only be accessed by a Galaxy Administrator. Additionally, the initial content of a Data Manager’s output file contains a JSON dictionary with a listing of the Tool parameters and Job settings (i.e. they are a type of OutputParameterJSONTool, this is also available for DataSourceTools). There is no requirement for the underlying Data Manager tool to make use of these contents, but they are provided as a handy way to transfer all of the tool and job parameters without requiring a different command-line argument for each necessary piece of information.
The primary difference between a standard Galaxy Tool and a Data Manager Tool is that the primary output dataset of a Data Manager Tool must be a file containing a JSON description of the new entries to add to a Tool Data Table. The on-disk content to be referenced by the Data Manager Tool, if any, is stored within the extra_files_path of the output dataset created by the tool.
- The tool tag must have the attribute: tool_type=“manage_data”
<tool id="data_manager_fetch_genome_all_fasta" name="Reference Genome" version="0.0.1" tool_type="manage_data">- The tool output must have format=“data_manager_json”
<outputs>
<data name="out_file" format="data_manager_json"/>
</outputs>- create
tools/data_manager/bwa_index_builder.xml- http://toolshed.g2.bx.psu.edu/repos/devteam/data_manager_bwa_index_builder/file/367878cb3698/data_manager/bwa_index_builder.xml
- Needs requirements tag for bwa application
- input params:
- a select param with options from_data_table=“all_fasta”
- a sequence_name text param
- a sequence_id text param
- outputs data param must be format
- transcribe the xml manually
- or you can download the file from: http://toolshed.g2.bx.psu.edu/repos/devteam/data_manager_bwa_index_builder/raw-file/367878cb3698/data_manager/bwa_index_builder.xml
- create
tools/data_manager/bwa_index_builder.py
- http://toolshed.g2.bx.psu.edu/repos/devteam/data_manager_bwa_index_builder/file/367878cb3698/data_manager/bwa_index_builder.xml
- Configure Galaxy to use this Data Manager tool
- add a data_manager entry inside data_managers tag in
data_mananger_conf.xml
- add a data_manager entry inside data_managers tag in
<data_manager tool_file="data_manager/bwa_index_builder.xml" id="bwa_index_builder" version="0.0.1">
<data_table name="bwa_indexes">
<output>
<column name="value" />
<column name="dbkey" />
<column name="name" />
<column name="path" output_ref="out_file" >
<move type="directory" relativize_symlinks="True">
<!-- <source>${path}</source>--> <!-- out_file.extra_files_path is used as base by default --> <!-- if no source, eg for type=directory, then refers to base -->
<target base="${GALAXY_DATA_MANAGER_DATA_PATH}">${dbkey}/bwa_index/${value}</target>
</move>
<value_translation>${GALAXY_DATA_MANAGER_DATA_PATH}/${dbkey}/bwa_index/${value}/${path}</value_translation>
<value_translation type="function">abspath</value_translation>
</column>
</output>
</data_table>
</data_manager>- run installer data_manager to build bwa indexes for sacCer2
- did it work?
- why not?
- add the missing bwa dependency
mkdir /home/galaxy/Desktop/Data_Managers/galaxy/tool_dependencies/bwa/0.5.9/binmv /home/galaxy/Desktop/Data_Managers/galaxy/galaxy-central/tool-data/sacCer1/bwa_index/sacCer1/bwa /home/galaxy/Desktop/Data_Managers/galaxy/tool_dependencies/bwa/0.5.9/bin/
- rerun the Data Manager and confirm that it is working
17:10-17:30 Put the DataManager in the Toolshed
- review toolshed best practices
- separate repository for required applications ( tool_dependencies.xml )
- separate repository for required custom datatypes ( repository_dependencies.xml )
- Run your own ToolShed.
- Add an administrator to tool_shed_wsgi.ini using the same process as before.
- For your public username, use devteam.
- Open a new terminal
- type
sh run_tool_shed.sh - Access the ToolShed at http://localhost:9009
- Using the Admin interface, create 2 new categories: “Data Managers” and “Tool Dependency Packages”
- Add an administrator to tool_shed_wsgi.ini using the same process as before.
- BWA Dependency from Main toolshed:
- Cross ToolShed dependencies are not currently supported, so we will use the import/export feature.
- Export the ‘package_bwa_0_5_9’ package from http://toolshed.g2.bx.psu.edu.
- This will create a ‘capsule_toolshed.g2.bx.psu.edu_package_bwa_0_5_9_devteam_ec2595e4d313.tar.gz’ file that can be imported into another ToolShed.
- In your local ToolShed, import your freshly exported ToolShed capsule.
- Prepare your Data Manager Toolshed Package.
- There are several ways to populate ToolShed repositories. We will be using mercurial to add files. Uploading tarballs is also possible and a very common way to do this.
- Login in to the toolshed
- “Click Create new repository”
- Add Create Repository fields:
- Name: data_manager_bwa_index_builder
- Synopsis: Data Manager that builds BWA indexes
- Detailed description: Index a FASTA file using the Burrows-Wheeler algorithm and populate the bwa_index.loc file.
- Categories: “Data Managers”
- Click “Save”
- Copy the clone link: “http://devteam@tool_shed/repos/devteam/data_manager_bwa_index_builder”
- In Eclipse:
- File —> New —> Project
- Clone Existing Mercurial Repository
- Click Next
- Paste the clone link
- Enter your password
- Click Next
- Click Next
- Click Finish
- Populate the repository
- Required files:
- data_manager/bwa_index_builder.py
- data_manager/bwa_index_builder.xml
- data_manager_conf.xml
- tool_data_table_conf.xml.sample
- tool-data/all_fasta.loc.sample
- tool-data/bwa_index.loc.sample
- tool_dependencies.xml
- Required files:
<?xml version="1.0"?>
<tool_dependency>
<package name="bwa" version="0.5.9">
<repository name="package_bwa_0_5_9" owner="devteam" changeset_revision="f8687dc2392c" toolshed="http://localhost:9009"/>
</package>
</tool_dependency> **Be sure to change the changeset_revision to the proper value.**- Use mercurial
- hg add
- hg ci -m “Populate data manager tool”
- hg push
- Remove your locally installed Data Manager from “data_manager_conf.xml”.
- Install new data_manager into your galaxy from the toolshed
- Build a bwa index for C. brenneri Feb. 2008 (WUGSC 6.0.1/caePb2)
17:30-18:00 Other Example data managers
SnpEff - http://snpeff.sourceforge.net/SnpEff.html
SnpEff is a variant annotation and effect prediction tool. It annotates and predicts the effects of variants on genes (such as amino acid changes).
- Key Points
- A Galaxy tool should guide the user to make valid parameter choices
- The reference data can either be in a history dataset or as reference data from tool-data
- The Galaxy Data Manager framework is flexible
- The SnpEff data manager populates multiple .loc files
- The data_manager_snpEff_databases.xml populates data_table “snpeff_databases” which provides the options for data_manager_snpEff_download.xml
- Initial Challenges
- SnpEff used a config file to specify the path to genome references, defaulted to user home directory
- Not compatible with galaxy structure for toolshed install and data manager operation
- Need downloaded genome version for SnpEff tool tests
- The available genome versions are dynamic, so the options shouldn’t be hardcoded into a Galaxy Select Parameter
- SnpEff galaxy tool has extra params that depend upon the reference data
- Every genome reference has file: snpEffectPredictor.bin
- Some genome references may include regulation, motif, and nextProt annotations
- SnpEff used a config file to specify the path to genome references, defaulted to user home directory
# Prevotella bryantii B14 has only the genome reference
$ ls tool-data/snpEff/data/ADWO01
snpEffectPredictor.bin
# Mouse build also has some regulation annotations
$ ls tool-data/snpEff/data/GRCm38.75/
regulation_ES.bin regulation_MEF.bin regulation_NPC.bin
regulation_ESHyb.bin regulation_MEL.bin snpEffectPredictor.bin
# Human build also has more regulation annotations and motif and nextProt annotations
$ ls tool-data/snpEff/data/GRCh37.75
motif.bin regulation_GM06990.bin regulation_HSMM.bin regulation_IMR90.bin regulation_NHEK.bin
nextProt.bin regulation_GM12878.bin regulation_HUVEC.bin regulation_K562.bin snpEffectPredictor.bin
pwms.bin regulation_H1ESC.bin regulation_HeLa-S3.bin regulation_K562b.bin
regulation_CD4.bin regulation_HMEC.bin regulation_HepG2.bin regulation_NH-A.bin- Solutions
- Discussed Galaxy requirements with application developer Pablo Cingolani who graciously added:
- command line option “-dataDir path” to specify the path to genome reference data
- download genome on demand functionality in SnpEff application, which allowed test cases to run without a preinstalled genome reference
- Developed 3 options in the SnpEff tool for getting the genome reference data
- Download on demand
- great for tests, but a lot of overhead for large genomes, and no way to capture genome specific annotations
- Discussed Galaxy requirements with application developer Pablo Cingolani who graciously added:
<tool id="snpEff" name="SnpEff" version="3.6">
<description>Variant effect and annotation</description>
<inputs>
<conditional name="snpDb">
<param name="genomeSrc" type="select" label="Genome source">
<option value="named">Named on demand</option>
</param>
<when value="named">
<param name="genome_version" type="text" value="GRCh37.68" label="Snpff Version Name"/>
</when>
</conditional>
</inputs>
</tool> - Data manager
- most efficient for multiuser or multi history use
- data_manager_snpEff_download.py inspects the downloaded genome files searching for added regulation and annotation files:
def download_database(data_manager_dict, target_directory, jar_path,config,genome_version,organism):
data_dir = target_directory
(snpEff_dir,snpEff_jar) = os.path.split(jar_path)
args = [ 'java','-jar' ]
args.append( jar_path )
args.append( 'download' )
args.append( '-c' )
args.append( config )
args.append( '-dataDir' )
args.append( data_dir )
args.append( '-v' )
args.append( genome_version )
proc = subprocess.Popen( args=args, shell=False, cwd=snpEff_dir )
return_code = proc.wait()
if return_code:
sys.exit( return_code )
## search data_dir/genome_version for files
regulation_pattern = 'regulation_(.+).bin'
# annotation files that are included in snpEff by a flag
annotations_dict = {'nextProt.bin' : '-nextprot','motif.bin': '-motif'}
genome_path = os.path.join(data_dir,genome_version)
if os.path.isdir(genome_path):
for root, dirs, files in os.walk(genome_path):
for fname in files:
if fname.startswith('snpEffectPredictor'):
# if snpEffectPredictor.bin download succeeded
name = genome_version + (' : ' + organism if organism else *)
data_table_entry = dict(value=genome_version, name=name, path=data_dir)
_add_data_table_entry( data_manager_dict, 'snpeff_genomedb', data_table_entry )
else:
m = re.match(regulation_pattern,fname)
if m:
name = m.groups()[0]
data_table_entry = dict(genome=genome_version,value=name, name=name)
_add_data_table_entry( data_manager_dict, 'snpeff_regulationdb', data_table_entry )
elif fname in annotations_dict:
value = annotations_dict[fname]
name = value.lstrip('-')
data_table_entry = dict(genome=genome_version,value=value, name=name)
_add_data_table_entry( data_manager_dict, 'snpeff_annotations', data_table_entry )
return data_manager_dict
def _add_data_table_entry( data_manager_dict, data_table, data_table_entry ):
data_manager_dict['data_tables'] = data_manager_dict.get( 'data_tables', {} )
data_manager_dict['data_tables'][data_table] = data_manager_dict['data_tables'].get( data_table, [] )
data_manager_dict['data_tables'][data_table].append( data_table_entry )
return data_manager_dict- SnpEff uses from_data_table to get options for params: regulation and extra_annotations
<tool id="snpEff" name="SnpEff" version="3.6">
<description>Variant effect and annotation</description>
<inputs>
<conditional name="snpDb">
<param name="genomeSrc" type="select" label="Genome source">
<option value="cached">Locally installed reference genome</option>
</param>
<when value="cached">
<param name="genomeVersion" type="select" label="Genome">
<!--GENOME DESCRIPTION-->
<options from_data_table="snpeff_genomedb">
<filter type="unique_value" column="0" />
</options>
</param>
<param name="extra_annotations" type="select" display="checkboxes" multiple="true" label="Additional Annotations">
<help>These are available for only a few genomes</help>
<options from_data_table="snpeff_annotations">
<filter type="param_value" ref="genomeVersion" key="genome" column="0" />
<filter type="unique_value" column="1" />
</options>
</param>
<param name="regulation" type="select" display="checkboxes" multiple="true" label="Non-coding and regulatory Annotation">
<help>These are available for only a few genomes</help>
<options from_data_table="snpeff_regulationdb">
<filter type="param_value" ref="genomeVersion" key="genome" column="0" />
<filter type="unique_value" column="1" />
</options>
</param>
</when>
</conditional>
</inputs>
</tool> - From history
- SnpEff Download tool allows users to proceed without the Galaxy admin
- The genome specific options are captured in metadata of the custom dataytpe: “snpeffdb”
$ cat snpeff_datatypes/datatypes_conf.xml
<?xml version="1.0"?>
<datatypes>
<datatype_files>
<datatype_file name="snpeff.py"/>
</datatype_files>
<registration>
<datatype extension="snpeffdb" type="galaxy.datatypes.snpeff:SnpEffDb" display_in_upload="True"/>
</registration>
</datatypes> $ cat snpeff_datatypes/lib/galaxy/datatypes/snpeff.py
"""
SnpEff datatypes
"""
import os,os.path,re,sys,gzip,logging
import galaxy.datatypes.data
from galaxy.datatypes.data import Text
from galaxy.datatypes.metadata import MetadataElement
log = logging.getLogger(__name__)
class SnpEffDb( Text ):
"""Class describing a SnpEff genome build"""
file_ext = "snpeffdb"
MetadataElement( name="genome_version", default=None, desc="Genome Version", readonly=True, visible=True, no_value=None )
MetadataElement( name="regulation", default=[], desc="Regulation Names", readonly=True, visible=True, no_value=[], optional=True)
MetadataElement( name="annotation", default=[], desc="Annotation Names", readonly=True, visible=True, no_value=[], optional=True)
def __init__( self, **kwd ):
Text.__init__( self, **kwd )
def set_meta( self, dataset, **kwd ):
Text.set_meta(self, dataset, **kwd )
data_dir = dataset.extra_files_path
## search data_dir/genome_version for files
regulation_pattern = 'regulation_(.+).bin'
# annotation files that are included in snpEff by a flag
annotations_dict = {'nextProt.bin' : '-nextprot','motif.bin': '-motif'}
regulations = []
annotations = []
if data_dir and os.path.isdir(data_dir):
for root, dirs, files in os.walk(data_dir):
for fname in files:
if fname.startswith('snpEffectPredictor'):
# if snpEffectPredictor.bin download succeeded
genome_version = os.path.basename(root)
dataset.metadata.genome_version = genome_version
else:
m = re.match(regulation_pattern,fname)
if m:
name = m.groups()[0]
regulations.append(name)
elif fname in annotations_dict:
value = annotations_dict[fname]
name = value.lstrip('-')
annotations.append(name)
dataset.metadata.regulation = regulations
dataset.metadata.annotation = annotations
try:
fh = file(dataset.file_name,'w')
fh.write("%s\n" % genome_version)
if annotations:
fh.write("annotations: %s\n" % ','.join(annotations))
if regulations:
fh.write("regulations: %s\n" % ','.join(regulations))
fh.close()
except:
pass- SnpEff tool gets options for params: regulation and extra_annotations from the “snpeffdb” metadata:
<tool id="snpEff" name="SnpEff" version="3.6">
<description>Variant effect and annotation</description>
<inputs>
<conditional name="snpDb">
<param name="genomeSrc" type="select" label="Genome source">
<option value="history">Reference genome from your history</option>
</param>
<when value="history">
<param format="snpeffdb" name="snpeff_db" type="data" label="SnpEff Genome Version Data"/>
<!-- From metadata -->
<param name="extra_annotations" type="select" display="checkboxes" multiple="true" label="Additional Annotations">
<help>These are available for only a few genomes</help>
<options>
<filter type="data_meta" ref="snpeff_db" key="annotation" />
</options>
</param>
<param name="regulation" type="select" display="checkboxes" multiple="true" label="Non-coding and regulatory Annotation">
<help>These are available for only a few genomes</help>
<options>
<filter type="data_meta" ref="snpeff_db" key="regulation" />
</options>
</param>
</when>
</conditional>
</inputs>
</tool> - Toolshed repositories
- package_snpeff_3_6 - Installs the SnpEff application as a tool_dependency https://testtoolshed.g2.bx.psu.edu/view/jjohnson/package_snpeff_3_6
$ find package_snpeff_3_6
package_snpeff_3_6/tool_dependencies.xml- snpeff_datatypes - defines custom datatypes https://testtoolshed.g2.bx.psu.edu/view/jjohnson/snpeff_datatypes
$ find snpeff_datatypes
snpeff_datatypes/datatypes_conf.xml
snpeff_datatypes/lib/galaxy/datatypes/snpeff.py- snpeff - Galaxy user tools https://testtoolshed.g2.bx.psu.edu/view/jjohnson/snpeff
$ find snpeff
snpeff/readme.rst
snpeff/repository_dependencies.xml
snpeff/snpEff.xml
snpeff/snpEff_download.xml
snpeff/snpEff_macros.xml
snpeff/test-data/vcf_homhet.vcf
snpeff/tool-data/snpeff_annotations.loc.sample
snpeff/tool-data/snpeff_databases.loc.sample
snpeff/tool-data/snpeff_genomedb.loc.sample
snpeff/tool-data/snpeff_regulationdb.loc.sample
snpeff/tool_data_table_conf.xml.sample
snpeff/tool_dependencies.xml- data_manager_snpeff - manages SnpEff reference data https://testtoolshed.g2.bx.psu.edu/view/jjohnson/data_manager_snpeff
$ find data_manager_snpeff
data_manager_snpeff/data_manager/data_manager_snpEff_databases.py
data_manager_snpeff/data_manager/data_manager_snpEff_databases.xml
data_manager_snpeff/data_manager/data_manager_snpEff_download.py
data_manager_snpeff/data_manager/data_manager_snpEff_download.xml
data_manager_snpeff/data_manager_conf.xml
data_manager_snpeff/repository_dependencies.xml
data_manager_snpeff/tool-data/snpeff_annotations.loc.sample
data_manager_snpeff/tool-data/snpeff_databases.loc.sample
data_manager_snpeff/tool-data/snpeff_genomedb.loc.sample
data_manager_snpeff/tool-data/snpeff_regulationdb.loc.sample
data_manager_snpeff/tool_data_table_conf.xml.sample
data_manager_snpeff/tool_dependencies.xml