Development News Brief
On this page
Get Galaxy
new: % hg clone http://www.bx.psu.edu/hg/galaxy galaxy-dist
upgrade: % hg pull -u -r 949e4f5fa03a
What’s New
ISMB/ECCB, BOSC, and 3DSIG 2011 Slides
Slides for several Galaxy related talks from this year’s ISMB/ECCB 2011 meeting are now available online: ISMB-ECCB-BOSC 2011
Recent Publications
Blankenberg D, Taylor J, Nekrutenko A, The Galaxy Team. “Making whole genome multiple alignments usable for biologists.” Bioinformatics. 2011 Jul 19. [Epub ahead of print]
Afgan, E., Baker, D., Team, t. G., Nekrutenko, A. and Taylor, J. (2011). “A reference model for deploying applications in virtualized environments.” Concurrency and Computation: Practice and Experience. doi: 10.1002/cpe.1836
After a period of quiescence, the GalaxyProject.org web site has been updated to more accurately reflect what is going on in Galaxy. This is a great starting point for information about Galaxy.
Wiki
Galaxy has a new Wiki at http://galaxyproject.org/wiki/. This includes a centralized location for help and screencasts. If you haven’t already done so, please create a login and a page to describe yourself, and your involvement with Galaxy.
And there is now an updated Galaxy article on Wikipedia: http://en.wikipedia.org/wiki/Galaxy_%28computational_biology%29. Additions and clarifications welcomed!
Revised and New Content
- Learn: A hub page for resources on learning how to use Galaxy.
- News Page and RSS Feed: News items of interest to the Galaxy community
- Events: Comprehensive listing of upcoming (and past) Galaxy Events
- Get Involved and Outreach: How to get involved with Galaxy and how to spread the word.
- Public Galaxy Servers: A list of publicly available Galaxy servers. If you have one, please add it to the list. PublicGalaxyServers
Tool Shed
http://galaxyproject.org/wiki/Tool%20Shed
- Introduction
- Basic repository features: create repository, upload, browse and delete files
- Viewing or managing a repository
- Contacting the owner of a repository
- Uploading additional files
- The mercurial repository change log
- Uploading a new version of an existing tool
- Repository revisions and download-able tool versions
- Repository rating and reviews
Upcoming Events
-
UAB Research Computing Day
- http://docs.uabgrid.uab.edu/wiki/2011
- September 15-16, Birmingham, Alabama, United States
- Galaxy: A Web-based Platform for Accessible, Reproducible, and Transparent High-throughput Biology, Jeremy Goecks, and Galaxy Workshop
-
Genome Informatics Workshop @ Beyond the Genome 2011
- http://www.beyond-the-genome.com/program.html
- September 19, Washington DC, United States
- Transparent, accessible, reproducible analysis with Galaxy, James Taylor
-
AWS Genomics Event
- http://aws.amazon.com/genomicsevent
- September 22, Seattle, Washington, United States
- Developing distributed analysis pipelines with shared community resources using CloudBioLinux and CloudMan, Brad Chapman
Updated & Improved
Data
- New pair-wise alignments for panTro2: calJac3, canFam2, danRer7, equCab2, galGal3, hg19, mm9, ponAbe2, rheMac2
- New pair-wise alignments for panTro3: calJac1, canFam2, equCab1, hg17, hg19, mm9, ponAbe2, rn4, calJac3, danRer4, galGal2, hg18, mm8, monDom4, rheMac2
- Correction: panTro3 now available as a native database for the tool “Fetch Sequences -> Extract Genomic DNA”
Tools
- liftOver tool enhancement: new options for the reporting of multiple output regions, a useful feature for cross-species conversions
- RNA-seq
- Added tool wrapper for Trinity, the de novo RNA-seq transcript assembler http://trinityrnaseq.sourceforge.net
- Enable Cufflinks to work when setting metadata externally
- Rename and reorder Cuffdiff outputs for clarity
- Make Cuffcompare wrapper compatible with v1.0.3
- Make Cufflinks/compare/diff wrapper versions independent of tool versions and add comments that indicate tool versions supported by wrappers
- Add GTF-guide option to Cufflinks wrapper
- BWA modified to run directly on fastqillumina files (no Fastq Groomer required)
- Add FIMO tool, from MEME suite, available on Test (only)
- FASTQ Groomer updated to run on Color Space files that contain an extra leading quality score (for adapter base) by removing the extra leading quality score, e.g. files obtained from the SRA
Visualization (Trackster)
- New features
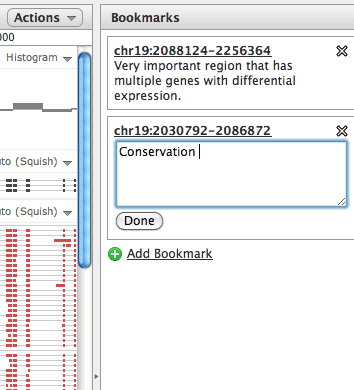
Bookmarking: image shows a bookmark being set for a genomic region.
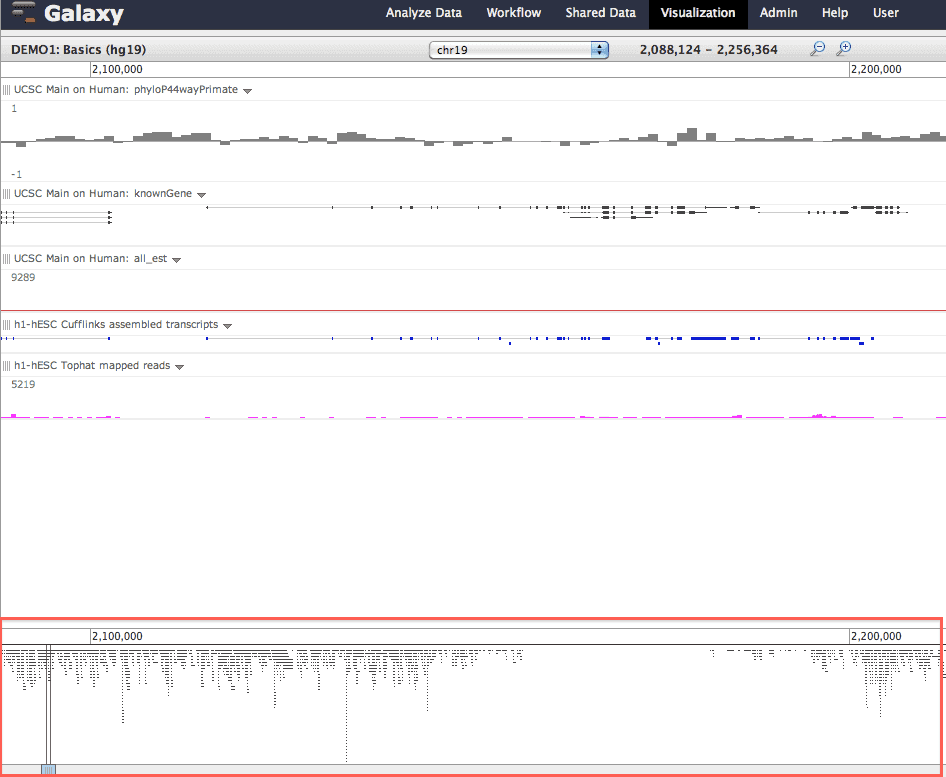
Overview tracks: image shows, at the bottom: (a) an overview for the EST track and (b) the region currently being viewed.
Features’ transparency based on attribute values: image shows transparency based on score, making it easy to identify a gene’s dominant isoform.
![]()
- Enhancements
- Enable filter chaining when running on complete dataset
- Add tracks in the order that they appear in the dataset listing
- Enable copying/importing of visualizations
- Provide option to fetch additional data as needed
- Show ‘Add Datasets’ button when there are no tracks
- Use requestAnimationFrame for better performance
- Tuning
- Additional security for loading visualizations
- Use error padding only when needed by placing error messages in div above tiles rather than in tiles
- Have child track inherit mode from parent track
- Bug fixes when (a) running tools in a shared visualization and (b) when drawing a track created by a tool
- Configuration
- New tag <trackster_conf> indicates that tool is compatible with Trackster
- Current tools compatible with Trackster include Cufflinks and GOPS intersect/subtract
Workflows
- Clone annotation and tags when cloning workflows
- Enable workflows to be uploaded to myExperiment
API
- API is now available for managing histories and datasets
- API now has the ability (for administrators and other defined users) to run a command as another user.
See API run_as for more details. - Expose workflows shared with the user in /workflows list of the API
- Addition of a case sensitivity notice for login not found error.
- Enhancements to handling format=“input” in workflows, fixes #582
- Added the ability for a user to use the API as another user via the setting ‘api_allow_run_as’ in the config file (thanks John Duddy!)
- Added the ability to create histories, delete histories, and create new history items from library items (thanks again John Duddy!)
Source
- UI Help -> Email link now points to http://galaxyproject.org/wiki/Support
- Enhancement code to support uploading various image data types (thanks Jelle Scholtalbers!)
- New collect_job_timings.sh script added to ~/contrib (thanks Assaf Gordon!)
- Several patches added to cleanup_datasets.py (thanks Assaf Gordon!)
- Update EuPathDB datasource configuration file
- Update GMAJ to latest version and fix exporting results from GMAJ back into Galaxy
- Added a generic XML datatype (thanks Peter Cock!)
- Added utility helper.py (previously posted to the galaxy-dev mailing list as “galaxythinger.py”)
- Command line tool for performing a couple common operations such as encoding/decoding IDs and getting the disk path of HDAs. This can be useful for system administration and debugging
Quotas
- User data and job quota limits are now implemented at the public Galaxy Test instance Test Quotas
- While no hard quotas are currently implemented at the public Galaxy Main instance, we do ask that users stay within certain usage limits: Main Quotas
- If you find that you require additional resources, please consider the alternative Galaxy options explained at: BigPicture/Choices
Bug Fixes
- Make message stating how much disk space improved in Galaxy reports, fixes #618
- Fix bug in rendering roles on the permissions page for data libraries (thanks Glen Beane!)
- In the data library hierarchy, rendered roles are all derived from roles associated with the LIBRARY_ACCESS permission, but roles rendered for that permission itself must be handled as a special case. Now, all legitimate roles not associated are listed.
- Fix for tool configs that do not include a command tag
- Fix a bug when setting default history permissions in the user preferences; receiving method now gets all required parameters
- Enable tags and annotations to work for new datasets in history;
- Reading and visualizing GFF3 datasets
- Removed unspecified build validators from Cufflinks/compare/diff tools as these cause trouble when running tools on custom genomes
- Disable update check in cuffdiff wrapper
- Disable grid controls and content when new content is being fetched, fixes #272
- Include post-job actions when importing and exporting workflows, fixes #518
- Improve filtering tool’s stdout reporting of lines filtered, fixes #536
- Show tool search by default; fixes #575
- Add support for global operations to grid framework; fixes #387
- Fix filtering of deleted objects in grid framework when using sqlite, also make history grid’s label for deleted filter clearer; fixes #596
- Remove unspecified build validators from Cuffdiff wrapper, fixes #631
- Remove unspecified build validator from extract_genomic_dna tool because it can work with a custom fasta
- Required fields for extra sample form data now enforced, fixes #301
- Proper inheritance in model for APIItem, fixes #527
- Administrative job lock toggling inappropriately. Split logical forms into actual separate forms, fixes #612
- Fix for rename not updating latest_workflow in addition to the stored workflow. This fixes the error where workflow exports always had the original workflow name, regardless of renames
- FTP uploads which contained a comma in the filename could not be imported unless importing more than one file at a time (thanks Ilya Chorny!)
- Fixed sending email when Sample Tracking Requests reach a terminal state
About Galaxy
The GalaxyTeam is a part of BX at Penn State, and the Biology and Mathematics and Computer Science departments at Emory University.
Galaxy is supported in part by NSF, NHGRI, the Huck Institutes of the Life Sciences, and The Institute for CyberScience at Penn State, and Emory University.
Join us at Twitter #usegalaxy
http://twitter.com/#!/search/galaxyproject