October 2013 Galaxy Update
On this page

Welcome to the October 2013 Galaxy Update, a monthly summary of what is going on in the Galaxy community. Galaxy Updates complement the Galaxy Development News Briefs which accompany new Galaxy releases and focus on Galaxy code updates.
New Public Servers
A record six new servers joined the list of over 40 publicly accessible Galaxy servers in September.
CNIC.DarwinTree
CNIC.DarwinTree is a molecular data analysis and application environment for DarwinTree, GSQCS, iDNAbar, NANNO, IPIP, and many other tools for phylogenetic analysis.
CNIC.DarwinTree is supported by the Chinese Academy of Sciences Computer Network Information Center (中国科学院计算机网络信息中心), and has [Email support](mailto:support AT cnic DOT cn) as well.
Fast UniFrac

Fast UniFrac provides a suite of tools for the comparison of microbial communities using phylogenetic information. See “Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data” by M Hamady, C Lozupone and R Knight, The ISME Journal (2010) 4, 17–27; doi:10.1038/ismej.2009.97
No login is required to use Fast UniFrac and the site has [email support](mailto:MicrobiomeHelp AT colorado DOT edu). Fast UniFrac is sponsored by the Knight Lab at the University of Colorado at Boulder.
kmer-SVM
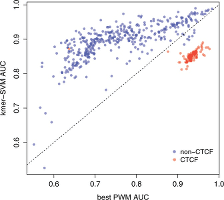
kmer-SVM is “a tool suite designed to aid in analysis of next-generation sequencing (NGS) data. Our suite uses a support vector machine (SVM) with kmer sequence features to identify predictive combinations of short transcription factor binding sites which determine the tissue specificity of the original NGS assay. Information gained from kmer-SVM can be used as an additional source of confidence in genomic experiments by recovering known binding sites, and can also reveal novel sequence features and possible cooperative mechanisms to be tested experimentally.”
The kmer-SVN server is describeded in “kmer-SVM: a web server for identifying predictive regulatory sequence features in genomic data sets.” Christopher Fletez-Brant; Dongwon Lee; Andrew S. McCallion; Michael A. Beer Nucleic Acids Research 2013; doi: 10.1093/nar/gkt519
A tutorial on using the web server and a Galaxy Tool Shed repository are also available. [Email support](mailto:kmersvm DOT team AT gmail DOT com) is provided, and the project is a collaboration between Christopher Fletez-Brant and Dongown Lee respectively of the McCallion Lab of the McKusick-Nathans Institute of Genetic Medicine at the Johns Hopkins University School of Medicine and the Beer Lab of the Johns Hopkins University Department of Biomedical Engineering.
NGS-QC Generator
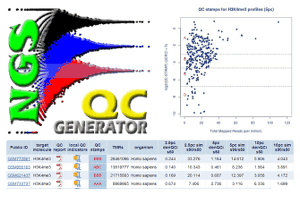
NGS-QC Generator evaluates the quality of ChIP-seq and enrichment-related NGS data using NGS-QC Generator, which also includes a database with preprocessed profiles and a tutorial on how to analyze sequencing profiles yourself.
See A quality control system for profiles obtained by ChIP sequencing by Mendoza-Parra, et al., in Nucl. Acids Res. (2013)
The server is hosted by the Gronemeyer lab and email support is available. You must have an account to use the server; anyone can create an account. “Due to storage space constraints, uploaded datasets into the Galaxy instance may not be available for more than 24hours, thus we strongly suggest users to download their processed files as early as possible.”
USP-FCFRP

The Laboratory of Biological Physics at USP-FCFRP Galaxy server has these tools:
ProtPred-GROMACS: Ab initio protein structure prediction framework which uses evolutionary algorithms to optimize objective functions such as potential energy, number of hydrogen bonds and solvent accessible surface area.
2PG: Faster and simpler algorithm in which one objective function is optimized
3PG: A more robust approach in which multiple objective functions are optimized at the same time
Protein Validation: Check the integrity of a PDB file
Protein Desolvation: Remove a solvation layer around your protein.
The Laboratory of Biological Physics, part of the Faculty of Pharmaceutical Sciences of Ribeirão Preto (FCFRP) at the University of São Paulo (USP). “You can use tools developed at our laboratory and by our collaborators for free. You just need to sign up to the server using your academic e-mail.”
ValidatorMAX

This server implements ValidatorMAX: confident identification of stable isotope-labeled peptide pairs and associated abundance ratios. The server is sponsored by the Department of Molecular Genetics and Cell Biology, Ludwig Center for Metastasis Research, Department of Pediatrics, Center for Research Informatics, Computation Institute, The University of Chicago
New Papers
| # | Tag | # | Tag | # | Tag | ||
|---|---|---|---|---|---|---|---|
| 2 | Cloud | 2 | RefPublic | - | UseLocal | ||
| 2 | HowTo | - | Reproducibility | 10 | UseMain | ||
| 4 | IsGalaxy | 1 | Shared | 1 | UsePublic | ||
| 21 | Methods | 8 | Tools | 1 | Visualization | ||
| - | Project | 2 | UseCloud | 14 | Workbench |
44 new papers were added to the Galaxy CiteULike Group in September. In addition to papers featuring the kmer-SVM and NGS-QC Generator (see above) these papers may be particularly interesting to the Galaxy community:
-
“Genomics in the clouds” by Vivien Marx, Nature Methods 10, 941–945 (2013)
-
“Galaxy tools and workflows for sequence analysis with applications in molecular plant pathology.” by Cock PJ, Grüning BA, Paszkiewicz K, Pritchard L. PeerJ 1:e167
-
“TALENoffer: genome-wide TALEN off-target prediction” by Jan Grau, Jens Boch, Stefan Posch, Bioinformatics (30 August 2013), doi:10.1093/bioinformatics/btt501
And the Galaxy Project now has it’s own CITATION file. CITATION files “tell readers how best to cite that software.” Also see the Citing Galaxy wiki page.
Who’s Hiring

The Galaxy is expanding! Please help it grow.
- Statistical Genomics Postdoc opening in the Makova lab at Penn State
- Computational biology opening at University Pierre-et-Marie-Curie, Paris
- M2 Développement et intégration d’outils pour la bioanalyse dans Galaxy, Limagrain à Chappes (Puy-de-Dôme, Auvergne)
- Part time position at GenePeeks
- Stage M2 Développement et intégration d’outils pour la bioanalyse dans Galaxy chez Vilmorin & Cie.
- PhD or postdoc position available at Laboratory of Computational Biology, University of Leuven
- The Galaxy Project is hiring software engineers and post-docs.
- Sr Bioinformatics Specialist, Tufts University, Boston MA.
Got a Galaxy-related opening? Send it to outreach@galaxyproject.org and we’ll put it in the Galaxy News feed and include it in next month’s update.
Events
### Australia!




In the next 3 months there are no less than 13 talks and workshops at at least 5 different venues on at least 7 distinct dates. Those 13 known events include 8 workshops. If you don't find some Galaxy training this spring then you aren't trying.
All Those Other Continents
And don’t worry, you won’t have to travel to Australia during its spring just to learn about Galaxy. There are also upcoming events in North America, Europe, and Africa.





Online Materials from Past Events


First, all videos from GCC2013 are now also available on Vimeo. They are still available on the Galaxy web site as well, but having them on Vimeo allows you to do all the things an online video provider supports, such as automatically embedding and linking videos from popular web sites.
Two other resources (at least) became available in September:
-
Informatics on High Throughput Sequencing Data Workshop from Bioinformatics.ca now has slides & video online.
-
Course Materials for the 2013 UC Davis Bioinformatics Short Course: This course featured 5 great days of lecture and hands-on exercises. And, an Amazon Web Services AMI that includes all tools and data used in the course, is also available.
See the list of other tutorials on the Learn hub page for more.
Galaxy Distributions
The most recent Galaxy distribution was August 12.
A new version of CloudMan was released in July.
Tool Shed Contributions

There were many…
- naive_variant_caller: process aligned reads, produce VCF file containing per position variant calls
- barrnap: ribosomal RNA finder for Bacterial genome sequences
- prokka (and a second wrapper): rapid annotation of prokaryotic genomes
- coverage_report: Generate Detailed Coverage Report from BAM file
- mugsy: multiple whole genome aligner
- edena: a de novo short reads assembler
- edge_pro: efficient gene expression level estimation in prokaryotic genomes from RNA-seq
- glimmer: find genes in microbial DNA, especially bacteria, archaea, and viruses
- get_fasta_from_taxon: Get FASTA from NCBI taxonomy ID
- sniploid2: compare SNPs detected from a polyploid to SNPs derived from its parental genomes
- ssake: de novo assembly of millions of very short DNA sequences
- sspace: scaffolding pre-assembled contigs using paired-read data
- annovar: Functional annotation of genetic variants from high-throughput sequencing data
- merge_fna_qual: Merges 454 Fasta and Quality files into a Fastq file.
- ncbi_sra_toolkit: NCBI Sequence Read Archive toolkit utilities
- sql_tools and sparql_tools: In-memory SQL and SPARQL runners and related tools
- compute_motifs_frequency, ctd_batch, compute_motif_frequencies_for_all_motifs, indels_3way, t_test_two_samples, xy_plot, divide_pg_snp, draw_stacked_barplots, split_paired_reads, mutate_snp_codon, hgv_hilbertvis, hgv_fundo, categorize_elements_satisfying_criteria, microsatellite_birthdeath, multispecies_orthologous_microsats, fasta_nucleotide_changer, fasta_clipping_histogram, fasta_formatter, fastq_quality_boxplot, fastq_quality_converter fastq_quality_filter fastq_to_fasta, and the fastx_toolkit all from devteam.