September 2014 Galaxy Update
On this page

Welcome to the September 2014 Galaxy Update, a summary of what is going on in the Galaxy community. Galaxy Updates complement the Galaxy Development News Briefs which accompany new Galaxy releases and focus on Galaxy code updates.
Galaxy-UK Community Launched
Galaxy-UK, a new Galaxy Community was launched in August. The Galaxy-UK Community aims to:
- Bring the Galaxy community in the United Kingdom closer together
- Identify and address the needs of the community
- Encourage interaction and collaboration.
Galaxy-UK is also an information hub for events such as:
- UK based Galaxy training courses
- UK based talks involving Galaxy
- Information on the location of UK Galaxy servers
- Anything else that might be pertinent to bring the UK Galaxy users/admins/trainers together as a community
The community will also have both online meetings and physical meetings, so keep an eye open for these events.
Other Galaxy Communities
Don’t fret if you want to join a community but you are not in the UK. Galaxy-UK is just one of several Galaxy communities you can join. (And there are rumors of a German language community in the works.)
Events
Galaxy Events in Europe, Fall 2014
There are a wealth of Galaxy related events in Europe this fall. Events include a large Galaxy presence at ECCB’14, the Fourth Galaxy User Group Grand Ouest (GUGGO) meeting, the 2014 Swiss-German Galaxy Tour, and several training events in Italy, the United Kingdom, Croatia, Norway, and France.
These events are a great way to meet other Galaxy users and developers and learn and share best practices. If you’re in Europe and are interested in learning more about Galaxy and/or the community, then please give these a look.



The Great GigaScience and Galaxy Workshop



The The Great GigaScience and Galaxy (G3) Workshop will be held, Friday 19 September 2014 at The University of Melbourne from 8:45-5pm.
The day’s theme is Turning data—big data—into research impact
Morning Session of Talks (8:45am - 12:00pm)
- Theatre 1, 207 Bouverie Street, Parkville.
- Featuring talks from David Vaux, A Speaker on Galaxy, Representative from NHMRC on open access and the GigaScience Editorial Team.
Afternoon Workshops (2:00pm - 5:00pm)
- Workshop Stream 1 - VLSCI Boardroom, 187 Grattan Street, Parkville
- Setting up your own personal galaxy instance and basic galaxy usage. Galaxy 101.
- Workshop Stream 2 - B117, 207 Bouverie Street, Parkville
- Authorea software carpentry workshop
See the event page for registration and contact links, and additional information.
Other Events
And don’t worry, Europe does not have a complete lock on upcoming Galaxy related events. There is also things going on in North America, and a few more in Australia too. See the Galaxy Events Google Calendar for details on other events of interest to the community.
| Date | Topic/Event | Venue/Location | Contact |
|---|---|---|---|
| October 29 - November 4 | Computational & Comparative Genomics Course Application Deadline: July 15 |
Cold Spring Harbor Laboratories (CSHL), New York, United States | William Pearson, Lisa Stubbs |
| November 2-5 | Introduction to Bioinformatics Analysis with Galaxy Workshop | ASA, CSSA, and SSSA International Annual Meeting, Long Beach, California, United States | Galaxy Outreach |
| SNP/Variant Analysis with Galaxy Workshop | |||
| A Gentle Introduction to Cloud Computing: Setting up your own Galaxy Server Workshop | |||
| November 19-20 | Workshop: Extended RNA-Seq analysis | The University of Queensland, Brisbane, Queensland, Australia | Mark Crowe |
| 2015 | |||
| July 6-8 | 2015 Galaxy Community Conference (GCC2015) | The Sainsbury Lab, Norwich, United Kingdom | Galaxy Outreach |
New Papers
44 papers were added to the Galaxy CiteULike Group in August, including this one:
-
Using galaxy-P to leverage RNA-Seq for the discovery of novel protein variations, by Gloria Sheynkman, James Johnson, Pratik Jagtap, et al.; BMC Genomics, Vol. 15, No. 1. (22 August 2014), 703, doi:10.1186/1471-2164-15-703
-
Integrating UIMA with Alveo, a human communication science virtual laboratory, by Dominique Estival, Steve Cassidy, Karin Verspoor, Andrew MacKinlay, and Denis Burnham; Proceedings of the Workshop on Open Infrastructures and Analysis Frameworks for HLT, pages 12–22, Dublin, Ireland, August 23rd 2014.
The new papers were tagged in many different areas:
| # | Tag | # | Tag | # | Tag | # | Tag | |||
|---|---|---|---|---|---|---|---|---|---|---|
| - | Cloud | - | Project | 1 | Tools | 6 | UsePublic | |||
| 2 | HowTo | - | RefPublic | - | UseCloud | 1 | Visualization | |||
| 1 | IsGalaxy | - | Reproducibility | 5 | UseLocal | 8 | Workbench | |||
| 30 | Methods | 1 | Shared | 8 | UseMain |
Who’s Hiring

The Galaxy is expanding! Please help it grow.
- Galaxy Workflow Developer, John Innes Centre, Norwich UK. Applications Close: 12 Sept
- Computational Science Developer I, Cold Spring Harbor Laboratory (CSHL), New York, United States
- Galaxy Platform Development Officer, The Genome Analysis Centre (TGAC), Norwich, United Kingdom. Closes 18 September.
- Bioinformatician position at Xenbase, University of Calgary.
- Statistical Genomics Postdoc opening in the Makova lab at Penn State
- The Galaxy Project is hiring software engineers and post-docs
Got a Galaxy-related opening? Send it to outreach@galaxyproject.org and we’ll put it in the Galaxy News feed and include it in next month’s update.
New Releases
August was an eventful month for releases. New versions of Galaxy, CloudMan, BioBlend, and blend4j were all released.
August 11, 2014 Galaxy Distribution

**[Complete News Brief](http://wiki.galaxyproject.org/DevNewsBriefs/2014-08-11)**
*Highlights:*
• [Security alert](http://tinyurl.com/nhgmbc5) from July 31st, upgrade now
• [Citations](http://wiki.galaxyproject.org/Admin/Tools/ToolConfigSyntax#A.3Ccitations.3E_tag_set): DOIs, `BibTeX`, and much much more
• [Docker](http://wiki.galaxyproject.org/Admin/Tools/Docker): You voted, we've *got it*, with a little help from our friends (you!)
• Significant Workflow, API, Job, Tool Shed, and Dataset management updates
• Fixes, tunings, plus just a drop of → Gossip
 |
getgalaxy.org | |
| galaxy-dist.readthedocs.org | ||
| bitbucket.org/galaxy/galaxy-dist | ||
| new: | $ hg clone https://bitbucket.org/galaxy/galaxy-dist#stable |
|
| upgrade: | $ hg pull $ hg update latest_2014.08.11 |
|
August 2014 CloudMan Release

This is mostly an incremental bug fix release with the following summary of changes:
- On AWS, updated galaxyFS snapshot (snap-e6e1c04a), which includes the June 2, 2014 Galaxy release with the July 30th security fix. All the tools installed via the Tool Shed have been updated and a number of new tools added, most notably: Tophat2, Bowtie2, FastQC, several FASTQ manipulation tools, several QC tools.
- For AWS, added support for VPC
- For OpenStack clouds, added the ability to automatically recover worker instances on cluster reboot
- Added support for creating a file system based on a downloadable archive
- Do not run Galaxy with multiple processes by default. This is because Tool Shed installs do not work properly in the multi-process mode. This feature can be enabled by setting user data option
configure_multiple_galaxy_processestoTruewhen launching an instance. - Set SGE slots in each queue to be equal to the number of cores on the instance
- Set instance IP in the Galaxy’s FTP data upload tool message
- Added support for Nginx v1.4 and allow it (with the PAM module) to used as the authentication mechanism when accessing Galaxy Reports app
- Fixed cluster deletion when performed via the API
- No longer automatically start Hadoop and HTCondor services
- On manually-invoked instance reboots, do not increment the instance reboot count that otherwise eventually leads to instance termination
- Limit the size of the log message buffer used in the UI to 1000 lines. Long-running instances had issues with this log growing large and that led to poor UI performance. The complete log is still available from the Admin page (or the command line).
- Automatically delete the bucket/container for Test type (i.e, ‘SGE only’) clusters on cluster termination
For complete details on implemented changes, please see the source code commits.
CloudMan offers an easy way to get a personal and completely functional instance of Galaxy in the cloud in just a few minutes, without any manual configuration.
BioBlend 0.5.1 Release
BioBlend version 0.5.1 was released on August 19. From the CHANGELOG:
- Fixed url joining problem described in issue #82
- Enabled Travis Continuous Integration testing
- Added script to create a user and get its API key
- Deprecated
create_user()method in favor of clearercreate_remote_user(). Addedcreate_local_user(). - Skip instead of fail tests when
BIOBLEND_GALAXY_URLandBIOBLEND_GALAXY_API_KEYenvironment variables are not defined. - Added export and download to objects API
- Added export/download history
- GalaxyClient: changed
make_put_requestto return wholerequestsresponse object - Added Tool wrapper to
BioBlend.objectsplus methods to list tools and get one - Added
show_tool()method toToolClientclass - Added
name,in_panelandtracksterfilters toget_tools() - Added
upload_dataset()method toHistoryclass. - Removed
DataInputandToolclasses for workflow steps.Toolis to be used for running single tools.
blend4j 0.1.1 Release
blend4j version 0.1.1 was released on August 27th. Some key features from the CHANGELOG:
- Dataset collection support by Aaron Petkau. Among other things the histories client can now create and return information about collections and the workflows client can specify dataset collections as inputs.
- Documentation overhaul - API documentation is now available online.
- Update tool shed client defaults to reflect the fact main tool shed is now being served over HTTPS.
Galaxy IPython

And … Björn Grüning and Helena Rasche also released Galaxy IPython:
We proudly present the first release of the Galaxy IPython project.
Galaxy IPython is a visualization plugin which should enable Galaxy users with coding skills to easily process their data in the most flexible way. With this plugin, it is possible to analyse and post-process data without downloading datasets or entire histories. One of our aims was to make Galaxy more attractive and accessible to bioinformaticians and programmers, and we hope that this project will build some bridges.
Disclaimer: Even though the Ipython notebooks can be stored and reused, this plugin will break the Galaxy philosophy of reproducibility, I feel personally bad about that, but I also think it is a great opportunity to get more bioinformaticians into Galaxy, and to get Galaxy used more often as a teaching resource. By being able to teach not only about workflows but also about data analysis tasks often necessary with Bioinformatics, Galaxy will be significantly more useful in teaching environments.
Keep in mind to write a nice Tool Shed Tool if you catch yourself using IPython in Galaxy to often for the same task.
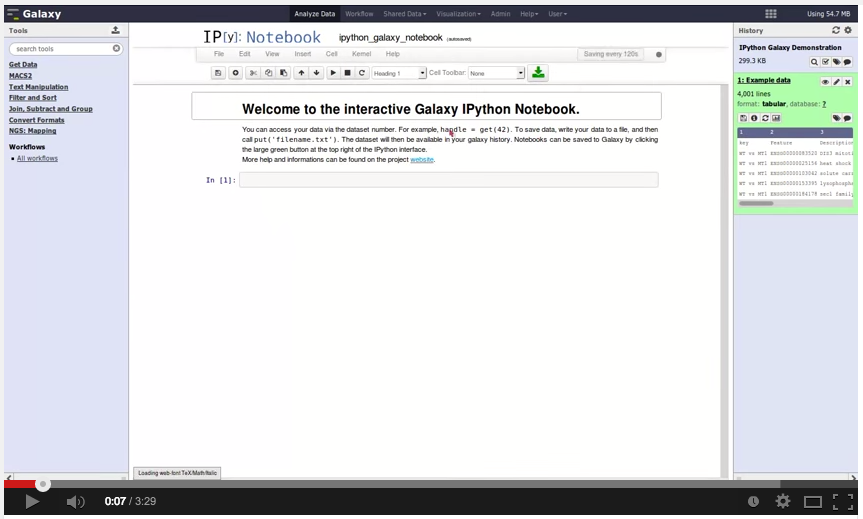
A few features we have up and running:
- Use IPython directly in the main window or in the Scratchbook
- Completely encapsulated IPython environment with matplotlib, biopython, pandas and friends already installed.
- IPython runs completely self-contained within a Docker container, separate from your Galaxy data
- Easy access to datasets from your current history via pre-defined IPython functions
- Manipulate and plot data as you like and export your new files back into your Galaxy history
- Save IPython Notebooks across analysis sessions in your Galaxy history with the click of a button.
- View saved IPython Notebooks directly in HTML format, or re-open them to continue your analysis.
- Self-closing and self-cleaning IPython Docker container
- Notebooks are secure, only accessible to the intended user
Please follow the installation instruction on our project page.
The underlying IPython Notebook (+Galaxy sugar) is stored at Github and the Docker Registry.
You can also install a ipynb datatype:
Comments welcome!
Happy research!
Eric & Björn
Galaxy Community Hubs


| Share your experience now |
There were no new Log Board or Deployment Catalog entries in August! Eek! Please don’t let this happen again!
The Community Log Board and Deployment Catalog Galaxy community hubs were launched last your. If you have a Galaxy deployment, or experience you want to share then please publish them this month.

ToolShed Contributions
Galaxy Project ToolShed Repos
Here are new contributions for the past two months.
In no particular order:
Tools
-
From lparsons
- sam_stats: generates basic sam/bam stats (from ea-utils package)
- fastq_join: merge overlapping paired-end reads (from ea-utils package)
-
From saskia-hiltemann
- ireport: create interactive HTML reports from galaxy outputs.
-
From iuc
- stringtie: fast and highly efficient assembler of RNA-Seq alignments into potential transcripts.
- vt_variant_tools: VT: a variant tool set that discovers short variants from Next Generation Sequencing data.
- bcftools: Utilities for variant calling and manipulating VCFs and BCFs
- gemini: GEMINI: a flexible framework for exploring genome variation
-
From urgi-team
- tandem_repeats_finder: locate and display tandem repeats in DNA sequences
-
From bgruening
- antismash: AntiSMASH - rapid genome-wide identification, annotation and analysis of secondary metabolite biosynthesis gene clusters
-
From agordon:
- datamash_wrapper: grouping and summarizing tool on tabular data files
-
From david-hoover:
- mirdeep2_and_targetspy_dh: Finding miRNA in NGS data and finding the targets for those miRNA
-
From big-tiandm:
- mirplant2: plant microRNA analysis tools
-
From devteam:
- correlation: computes the matrix of correlation coefficients between numeric columns.
- vcf2pgsnp: Convert from VCF to pgSnp format
- pgsnp2gd_snp: onverts a pgSnp dataset to gd_snp format, either starting a new dataset or appending to an old one.
- ucsc_custom_track: Build custom track for UCSC genome browser
- dna_filtering: Filter on ambiguities in polymorphism datasets
- mine: Applies the Maximal Information-based Nonparametric Exploration strategy to an input dataset.
- pearson_correlation: Computes Pearson’s correlation coefficient between any two numerical columns. Column numbers start at 1.
- generate_pc_lda_matrix: generate a matrix to be used for running the Linear Discriminant Analysis as described in Carrel et al., 2006 (PMID: 17009873)
- count_gff_features: Counts the number of features in a GFF dataset.
- column_maker: computes an expression for every row of a dataset and appends the result as a new column (field).
- scatterplot: creates a simple scatter plot between two variables containing numeric values of a selected dataset.
- plot_from_lda: generates a Receiver Operating Characteristic (ROC) plot that shows LDA classification success rates for different values of the tuning parameter tau as Figure 3 in Carrel et al., 2006 (PMID: 17009873).
- histogram: computes a histogram of the numerical values in a column of a dataset.
- lda_analysis: Perform Linear Discriminant Analysis
- snpfreq: basic analysis of bi-allelic SNPs in case-control data, using the R statistical environment and Fisher’s exact test to identify SNPs with a significant difference in the allele frequencies between the two groups
-
From arkarachai-fungtammasan:
- microsatellite_ngs: Pipeline to profile and genotype microsatellites from short read data
-
From peterjc:
- blast_rbh: BLAST Reciprocal Best Hits (RBH) from two FASTA files
-
From nikos:
- bigwig_to_wig: Converts a bigWig file to Wiggle (WIG) format
-
*From mcharles:
-
*From alan-blakely:
- filter_by_substring_match: Allows for partial sequences to match lines containing a larger, more complete sequence.
-
From anton:
- bamtools_filter: Filter BAM files on a variety of attributes
Data Managers
- From iuc:
- data_manager_gemini_database_downloader: Manage GEMINI databases. This tool will retrieve all files for the use in GEMINI.
Suites
- From anton
- suite_vcflib_tools_2_0: tools for manipulation of VCF files
Packages / Tool Dependency Definitions
-
From iracooke
- package_peptideshaker_0_31: Installs tool dependencies for PeptideShaker 0.31
- package_searchgui_1_19: Installs tool dependencies for !SearchGUI 1.19
-
From https://toolshed.g2.bx.psu.edu/view/anton
- vcflib_86723982aa: Compiled binary files for vcflib toolkit 86723982aa
- package_bamtools: bamtools - a collection of utilities for processing of bam files
-
From lparsons
- package_ea_utils_1_1_2_484: Command-line tools for processing biological sequencing data. Barcode demultiplexing, adapter trimming, etc. See ea-utils.
- package_rseqc_2_3_9: downloads and compiles version 2.3.9 of RSeQC.
-
From iuc
- package_stringtie_0_97:
- package_vcftools_0_1_12b: downloads and compiles version 0.1.12b of the vcftools suite.
- package_vt_5c735ab14b5603d9f14da6ee0e63d86ba3779934: a variant tool set that discovers short variants from Next Generation Sequencing data.
- package_htseq_0_6: downloads and compiles version 0.6 of the htseq package.
- package_pandas_0_14: downloads and compiles version 0.14 of the python library pandas, an open source, BSD-licensed library providing high-performance, easy-to-use data structures and data analysis tools for the Python programming language.
- package_bedtools_2_19: downloads and compiles version 2.19 of bedtools, a swiss army knife for genome arithmetic.
- package_grabix_0_1_3: downloads and compiles version 0.1.3 of grabix. grabix leverages the fantastic BGZF library in samtools to provide random access into text files that have been compressed with bgzip
- package_gemini_0_10_0: downloads and compiles version 0.10.0 of GEMINI. GEMINI (GEnome MINIng) is designed to be a flexible framework for exploring genetic variation in the context of the wealth of genome annotations available for the human genome.
- package_r_ggplot2_0_9_3: downloads and compiles verion 0.9.3.x from gglot2 the R package. ggplot2 is a plotting system for R, based on the grammar of graphics, which tries to take the good parts of base and lattice graphics and none of the bad parts.
-
From jjohnson:
- package_shear_0_2_11: Download and install SHEAR version 0.2.11 (Sample Heterogeneity Estimation and Assembly by Reference)
-
From agordon:
- package_datamash_1_0_5: grouping and summarizing tool on tabular data files
-
From devteam:
- package_mine_1_0_1: downloads and installs version 1.0.1 of the MINE .jar package.
Other News
- Basic variant calling in Galaxy online tutorial by Cynthia Gibas at Genome Intelligence
- From ron Horst: You can now BLAST search DNA & Proteins on galaxy-qld.genome.edu.au, using BRAEMBL resources. Coming soon to more Gernomics Virtual Lab VMs